
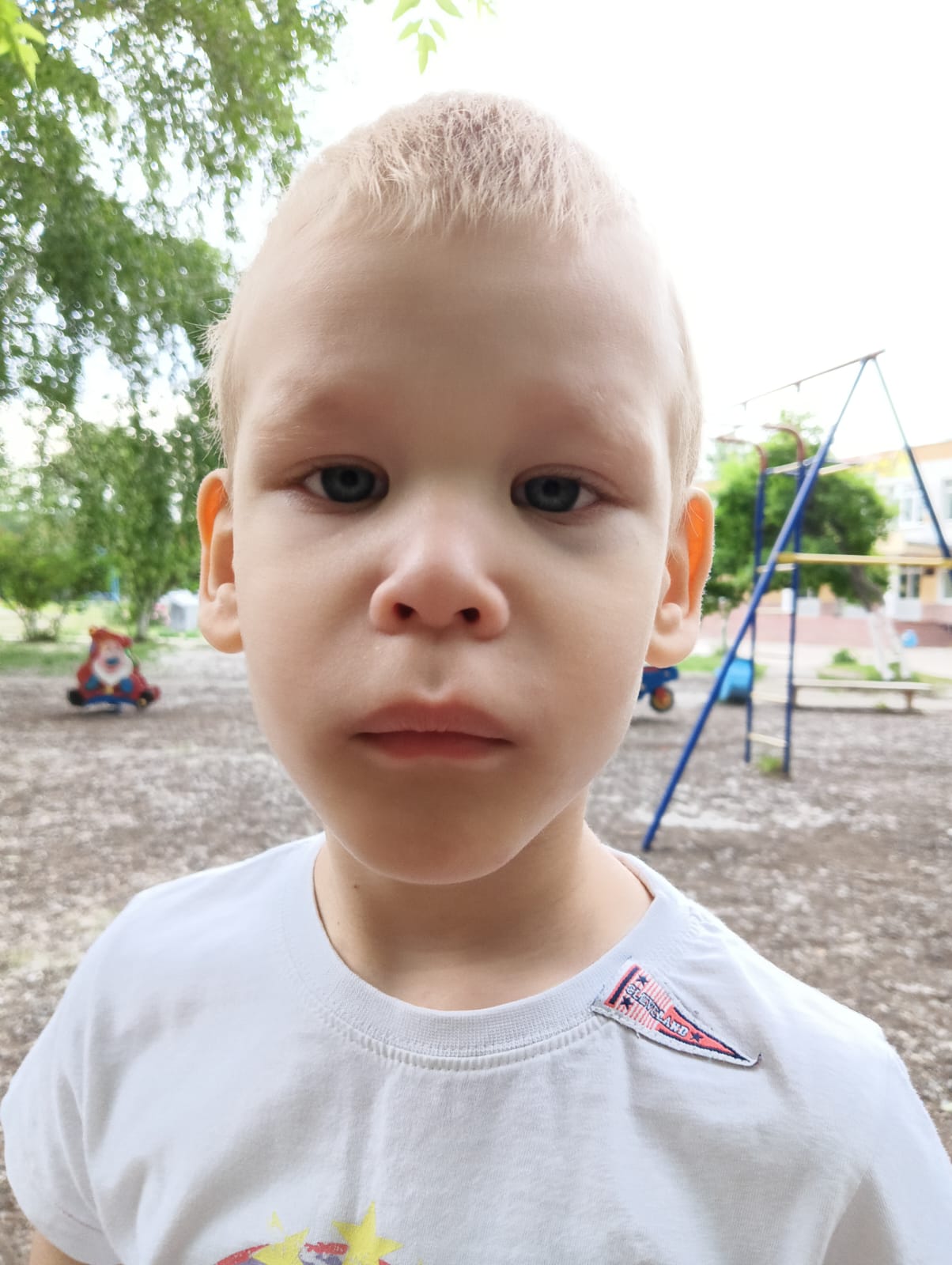


Российское сообщество родителей детей с синдромом Фелан-МакДермид


О проекте
Если в вашей семье кому-то поставлен диагноз синдром Фелан-МакДермид (ФМД), знайте – вы не одни!
Мы объединяемся для помощи детям и взрослым с синдромом Фелан-МакДермид. В нашем сообществе состоят семьи не только из России, но и из других стран.
О проекте
Синдром Фелан-МакДермид – очень редкое генетическое заболевание, ассоциированное с микроделецией хромосомы 22q13 или точечными мутациями на конце длинного плеча хромосомы 22 в гене SHANK3.
Впервые синдром был описан врачами Кэти Фелан и Хизер МакДермид в 1985 году.
По данным нашего сообщества в России около 70 детей с данным синдромом, но мы уверены, что таких семей гораздо больше, так как имеются проблемы с диагностикой данного синдрома ввиду недостатка информации о заболевании в медицинском сообществе, а также потому, что у этого синдрома есть схожие симптомы с другими РАС – по этой причине многим детям ставят диагноз РАС без генетического анализа.
Симптомы синдрома очень разнообразны и зависят от степени изменений гена SHANK3. В нашем сообществе есть дети с разными вариантами нарушений гена SHANK3, а именно: делецией, мутацией и несколько случаев дупликации гена.
Дети с делецией в гене SHANK3 чаще остальных имеют более выраженное отставание в развитии, как в физическом, так и умственном. У нескольких детей с делецией развита речь и способность к коммуникации, у остальных детей с делецией речь отсутствует, нет навыков коммуникации и практически полностью отсутствует понимание обращенной речи. Возможно такое отличие в развитии детей с делецией гена SHANK3 связано с размером делеции и местом ее расположения в гене.
Дети с мутацией в гене SHANK3 более перспективны в развитии и коммуникации, способность к развитию речи у таких детей выше, но по нашей статистике у них более выражены симптомы аутизма.
Интересным остается тот факт, что дети нашего сообщества, имеющие нарушения еще и в ряде других генов, помимо гена SHANK3, развиваются лучше, чем те, у кого задет только один SHANK3.
Как такового лечения синдрома Фелан-МакДермид в настоящее время не существует. Осведомленность о данном синдроме в медицинском сообществе очень низкая. В ряде зарубежных стран имеются разработки препаратов для данного синдрома, в том числе генного препарата (на стадии клинических исследований), на который мы возлагаем большие надежды.
Одна из основных целей данного проекта, созданного нашим сообществом Фелан-МакДермид — привлечение внимания научно-исследовательских центров в России и других странах, а также медицинского сообщества, для разработки необходимых методов лечения наших детей, с использованием опыта зарубежных специалистов, а также регистрация и применение в России препаратов, способных помочь детям с синдромом Фелан-МакДермид.
Мы приглашаем к сотрудничеству врачей, научных сотрудников федеральных медицинских учреждений различного уровня, научных сотрудников и представителей исследовательских центров, представителей власти, депутатов, социально ориентированные некоммерческие организации, фонды, а также СМИ для освещения синдрома Фелан-МакДермид.
Вся информация, представленная в данном разделе, описана с использованием статистических данных, собранных в результате опроса среди семей нашего сообщества, и не имеет рекомендательного характера.
Любое использование или копирование фотографий, видео, текста и иных материалов с данного сайта допускается только с разрешения правообладателя.
Сообщество выражает благодарность за помощь в создании сайта:







Авторы проекта: Анастасия Хоменко и Екатерина Симонова.
Наши цели:








Новости

23 сентября 2025 года. Исследование генной терапии JAG201 у детей и взрослых с гаплонедостаточностью SHANK3.
24 июня 2025 года. Регистрация МОО «ФМД-СООБЩЕСТВО» (Межрегиональная Общественная организация «Сообщество Родителей Детей с Синдромом Фелан-МакДермид», ОГРН 1255000059313).

20 марта 2025 года. На базе ФГБУ МГНЦ им. Бочкова создана рабочая группа для разработки клинических рекомендаций по синдрому Фелан-МакДермид. Ориентировочный срок подготовки проекта КР – декабрь 2025 года.

26 декабря 2024 года. Синдром Фелан-МакДермид включен в перечень орфанныхзаболеваний.
19 декабря 2024 года. Мы обратились на горячую линию с Президентом Российской Федерации В.В. Путиным.

Наши дети должны иметь право на социальную среду

Причины синдрома Фелан-МакДермид
23 сентября 2025 года. Исследование генной терапии JAG201 у детей и взрослых с гаплонедостаточностью SHANK3.
Исследователи генного препарата JAG201, который дает большие надежды семьям, столкнувшимся с синдромом Фелан-МакДермид, приступили к открытому исследованию фазы 1/2 с повышением дозы, для оценки безопасности, переносимости и клинической эффективности однократного введения JAG201 путем интрацеребровентрикулярной (ИЦВ) инъекции у детей и взрослых с гаплонедостаточностью SHANK3, вызванной мутациями, приводящими к потере функции SHANK3, и хромосомными делециями, охватывающими ген SHANK3. Клинические данные будут оцениваться с точки зрения безопасности, переносимости и предварительной клинической эффективности JAG201 у детей и взрослых с гаплонедостаточностью SHANK3. Сначала начнется набор детей, а набор взрослых может быть начат позже.
24 июня 2025 года. Регистрация МОО «ФМД-СООБЩЕСТВО» (Межрегиональная Общественная организация «Сообщество Родителей Детей с Синдромом Фелан-МакДермид», ОГРН 1255000059313).
Рады сообщить, что 24.06.2025 г. наше сообщество родителей детей с синдромом Фелан-МакДермид официально зарегистрировано в Министерстве юстиции Российской Федерации. Теперь все наши обращения в органы власти будут осуществляться официально от юридического лица. Регистрация сообщества расширяет возможности родителей детей с редким генетическим (орфанным) заболеванием при решении проблем, связанных с синдромом, дает возможность дополнительной информационной осведомленности о синдроме Фелан-МакДермид среди врачей, повышает шансы быть услышанными главой государства и федеральными органами исполнительной власти, в первую очередь — Министерством здравоохранения Российской Федерации. Наличие официально зарегистрированного сообщества может помочь ускорить обеспечение наших детей с редким генетическим заболеванием необходимыми препаратами, лечением, питанием и помочь в разработке индивидуальных методов реабилитации.
20 марта 2025 года. На базе ФГБУ МГНЦ им. Бочкова создана рабочая группа для разработки клинических рекомендаций по синдрому Фелан-МакДермид. Ориентировочный срок подготовки проекта КР – декабрь 2025 года.
Активной группе сообщества родителей детей с синдромом Фелан-МакДермид удалось встретиться с руководством ФГБУ МГНЦ им. Бочкова и обсудить возможность помощи нашим детям, в том числе — разработку клинических рекомендаций для данного синдрома. Клинические рекомендации очень важны для нас, так как в настоящее время мало кто из врачей знает о нашем синдроме, соответственно нет никаких рекомендаций по диагностике и лечению синдрома Фелан-МакДермид, хотя во многих зарубежных странах уже имеется ряд препаратов, способных уменьшить проявление симптомов, связанных с синдромом, и эффективно использующихся для лечения.
26 декабря 2024 года. Синдром Фелан-МакДермид включен в перечень орфанныхзаболеваний.
По результатам обращения на программу с Президентом Российской Федерации В.В.Путиным, синдром Фелан-МакДермид был включен в перечень орфанных заболеваний и ему присвоен код МКБ-10 Q93.5
https://minzdrav.gov.ru/documents/9837-perechen-redkih-orfannyh-zabolevaniy
19 декабря 2024 года. Мы обратились на горячую линию с Президентом Российской Федерации В.В. Путиным.
Актив сообщества и родители записали и отправили на программу «Итоги года с Владимиром Путиным 2024» видеообращение. Мы обратились к Президенту Российской Федерации В.В. Путину с просьбой о включении синдрома Фелан-МакДермид в перечень орфанных заболеваний, чтобы наши дети могли получать всю необходимую помощь, а также о содействии в актуализации проблемы генетических заболеваний в России и повышении информированности медицинского сообщества о синдроме Фелан-МакДермид и других генетических заболеваниях.
Знайте, что вы не одиноки!
